
Project: Conversion of IUPAC name of a molecule to its subsequent IUPAC chemical structure.
Platfrom: Ruby on Rails and OpenBabel (Rubabel)
Webpage: https://github.com/nicolejung/IUPAC
Summary and a brief algorithm to work on it:
Over many years, numerous recommendations for chemical nomenclature have been codified by the IUPAC (International Union of Pure and Applied Chemistry). There are several different approaches to representing chemical concepts that are in common use. Therefore, the system has to be able to use different vocabularies and different grammars to cope with these different styles of nomenclature. Moreover, multiple styles of nomenclature may be present within the same name. The removal of characters (elision) and the addition of characters are commonly employed to yield better sounding but semantically identical names. The recommendations contain many exceptions and irregularities due to their development by various chemists over time.
It’ll have a variety of conversions from conventional representations of various characters, e.g., Greek letters into a standard normalized representation. It has no initial preconception of how a chemical name should be broken down into words and further into tokens. A token is a string of characters that typically corresponds to a smallest part in a chemical name. The smallest part of a name that has semantic meaning, e.g., ‘eth’ in ‘ethene’ which means a carbon chain of length 2. The usage of white space and other English language token separators, such as hyphens, is quite different in chemical names from the usage in English language itself. The usage of these delimiters in chemical names is not always consistent, and some omissions or inclusions of separators may be regarded as errors by the IUPAC recommendations.
Chemical nomenclature can be thought of as being an artificial language. However, unlike other artificial languages, such as programming languages, the smallest past of chemical names are often not clearly delimited. One approach to this problem is to create all possible tokenization for a chemical name on the basis of the various tokens. However, as such an approach does not take into account context, and many tokenization will ultimately be found to be incorrect. For example ‘propan-2-ol’ would be tokenized to [‘prop’, ‘an’, ‘-’, ‘2-’, ‘ol’] and [‘propa’, ‘n-’, ‘2-’, ‘ol’], for which the latter is clearly wrong (the ‘n-’ here is equivalent to that found in ‘n-butane’).
One of the similar work done in this field is by Opsin designed and developed Unilever Centre for Molecular Science Informatics, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, England
They have produced an open source, freely available, algorithm (Open Parser for Systematic IUPAC Nomenclature, OPSIN) that interprets the majority of organic chemical nomenclature in a fast and precise manner. This has been achieved using an approach based on a regular grammar. This grammar is used to guide tokenization, a potentially difficult problem in chemical names. From the parsed chemical name, an XML parse tree is constructed that is operated on in a stepwise manner until the structure has been reconstructed from the name. Results from OPSIN on various computer generated name/structure pair sets are presented. These show exceptionally high precision (99.8%+) and, when using general organic chemical nomenclature, high recall (98.7−99.2%). This software can serve as the basis for future open source developments of chemical name interpretation.
In accordance to the OPSIN out project is quite similar and quite different as well. The language for the implementation by Opsin is done using the Java computing language and XML file but the platform we are using is Ruby. Ruby is a dynamic, reflective, object-oriented, general-purpose programming language. The syntax of Ruby is broadly similar to that of Perl and Python. Class and method definitions are signaled by keywords. One of the differences of Ruby compared to Python and Perl is that Ruby keeps all of its instance variables completely private to the class and only exposes them through accessor methods (attr_writer, attr_reader, etc.). It implements the concept of metaprogramming i.e. writing the whole code into one single line. This is where OPSIN and our project is different. But we share a similarity that we after the processing of the Chemical name of the compound. To convert it into the final structure or the .mol file we uses the OpenBabel. We are converting the name of the compound into the smiles strings so that it can be converted into the .mol or the 2D structure .
Smiles
The simplified molecular-input line-entry system (SMILES) is a specification in form of a line notation for describing the structure of chemical species using short ASCII strings. SMILES strings can be imported by most molecule editors for conversion back into two-dimensional drawings or three-dimensional models of the molecules.
Atoms are represented by the standard abbreviation of the chemical elements, in square brackets, such as [Au] for gold. Brackets can be omitted for the "organic subset" of B, C, N, O, P, S, F, Cl, Br, and I. All other elements must be enclosed in brackets. If the brackets are omitted, the proper number of implicit hydrogen atoms is assumed; for instance the SMILES for water is simply O.
Bonds between aliphatic atoms are assumed to be single unless specified otherwise and are implied by adjacency in the SMILES string. For example the SMILES for ethanol can be written as CCO. Ring closure labels are used to indicate connectivity between non-adjacent atoms in the SMILES string, which for cyclohexane and dioxane can be written as C1CCCCC1 and O1CCOCC1 respectively. Double, triple, and quadruple bonds are represented by the symbols '=', '#', and '$' respectively as illustrated by the SMILES O=C=O (carbon dioxide), C#N (hydrogen cyanide) and [Ga-]$[As+] (gallium arsenide).
Aromatic C, O, S and N atoms are shown in their lower case 'c', 'o', 's' and 'n' respectively. Benzene, pyridine and furan can be represented respectively by the SMILES c1ccccc1, n1ccccc1 and o1cccc1. Bonds between aromatic atoms are, by default, aromatic although these can be specified explicitly using the ':' symbol.
Branches are described with parentheses, as in CCC(=O)O for propionic acid and C(F)(F)F for fluoroform. Substituted rings can be written with the branching point in the ring as illustrated by the SMILES COc(c1)cccc1C#N.
Our project is a ruby gem and can be downloaded easily just by trying “gem install iupac” from the command line of Linux or can be downloaded from the github from https://github.com/nicolejung/IUPAC.
The tree line for our project can be shown below,
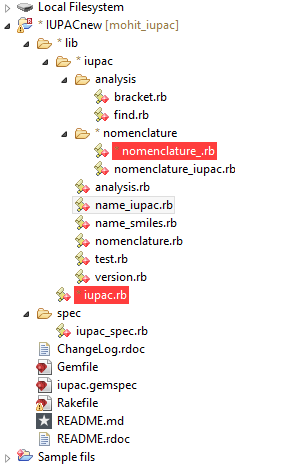
Our project is a ruby gem and hence all the directories are placed as a ruby application. In analysis folder we have two ruby files analysis/find.rb and analysis/bracket.rb. The find.rb has all the functions i.e. to find suffix, to find the parent chain, to find prefix, to find the double and triple bonds, to find the multiplier, to find the position of prefix and suffix or multipliers. The bracket.rb has the function to resolve the chemical name which has bracket and side chains in it.
In the folder nomenclature we have nomenclature.rb which has the all the nomenclature of the chemical names i.e. the smallest tokens or the chemical name which can be identified discreetly. Instead of using a files for different tokens(smallest chemical compounds) we use hash for storing these tokens into the file nomenclature,rb.
Then we have name_iupac.rb this is the function which we call different function recursively to process the chemical name. We’re working on the chemical name from right to left and simultaneously dropping the chunk of the molecule which has been detected.
In name_smiles.rb we create the smiles format of the processed chemical name.
And in test.rb we’ve taken the possible test cases of various IUPAC molecules and custom text settings.
To run the application we have to run iupac.rb file. And the prompt will ask you to whether you want to run the test cases or you want to insert the molecule by yourself.
Open Babel: The Open Source Chemistry Toolbox
Open Babel is a free, open-source version of the Babel chemistry file translation program. Open Babel is a project designed to pick up where Babel left off, as a cross-platform program and library designed to interconvert between many file formats used in molecular modeling, computational chemistry, and many related areas. Open Babel includes two components, a command-line utility and a C++ library. The command-line utility is intended to be used as a replacement for the original babel program, to translate between various chemical file formats. The C++ library includes all of the file-translation code as well as a wide variety of utilities to foster development of other open source scientific software.
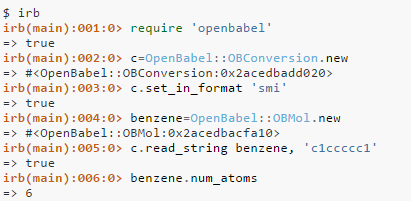
Open babel is then comes into the picture when we have a SMILES format of the molecule then it can convert it into a 2D structure of .mol file or any required file format we want. Open Babel is a project to facilitate the interconversion of chemical data from one format to another – including file formats of various types. This is important for the following reasons:
Multiple programs are often required in realistic workflows. These may include databases, modeling or computational programs, visualization programs, etc.Many programs have individual data formats, and/or support only a small subset of other file types.
Chemical representations often vary considerably:
Some programs are 2D. Some are 3D. Some use fractional k-space coordinates.
Some programs use bonds and atoms of discrete types. Others use only atoms and electrons.
Some programs use symmetric representations. Others do not.
Some programs specify all atoms. Others use “residues” or omit hydrogen atoms.
Individual implementations of even standardized file formats are often buggy, incomplete or do not completely match published standards. Open Babel improves this by allowing to access all kinds of file format easily.